
Studium
Chemie Diplom an der RWTH Aachen
Schwerpunkte: Organische und Makromolekulare Chemie.
Promotion
Totalsynthese von (+)-3-Oxacarbacyclin mittels asymmetrischer Olefinierungs- und Deprotonierungs-Reaktionen
Die Ergebnisse sind ausführlich in Liebigs Annalen 1997, 2419-2431, 2433-2441, Eur. J. Org. Chem. 1998, 805-826 oder in meiner Doktorarbeit „Totalsynthese von (+)-3-Oxacarbacyclin mittels asymmetrischer Olefinierungs- und Deprotonierungs-Reaktionen, Rainer Karl Ludwig Ossenkamp, RWTH Aachen, 1997 nachzulesen.
Auszug aus der Doktorarbeit:
Zusammenfassung
Ausgehend vom prochiralen Monoketon 29 erhielt man 3-Oxacarbacyclin (28) mit einer Gesamtausbeute von 1.5 % über 18 Stufen. Die wichtigen Syntheseschritte mit Ausbeuten und Selektivitäten sind in der Abb. 72 dargestellt.
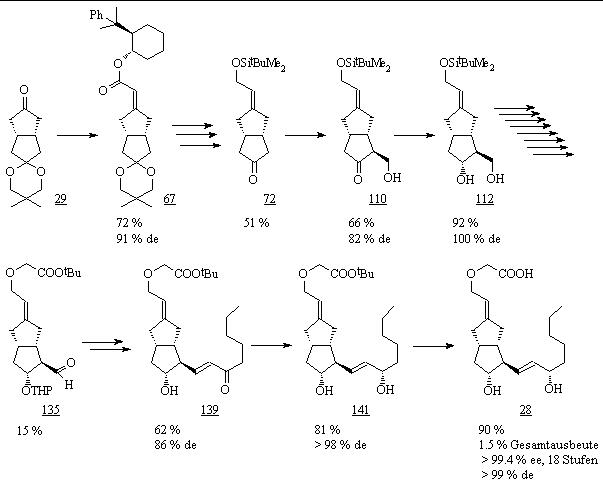
Abb. 72: Totalsynthese von 3-Oxacarbacyclin (28).
Ein wesentlicher Schwerpunkt bestand in der Optimierung der Stereoselektivität und Ausbeute der beiden maßgeblichen Schlüsselschritte dieser Totalsynthese – die asymmetrische HWE-Reaktion und die asymmetrische Deprotonierung zur stereoselektiven Einführung der a- und w-Seitenkette.
Dazu wurden 5 chirale Phosphonate dargestellt, mit denen man unter verschiedenen Reaktionsbedingungen HWE-Reaktionen mit dem Monoketon 29 durchführte (Abb. 73). Mit den Phosphonaten 43, 46 und 40 erhielt man das Olefin 59, mit dem Phosphonat 52 das Olefin 67 und mit dem Phosphonat 21 das Olefin 66, das bis auf eine zusätzliche CH3-Gruppe am Cyclohexanring dem Olefin 67 gleicht.
Zur Untersuchung der HWE-Reaktion wurden bei der Darstellung des Olefins 59 der Enantiomerenüberschuß und die Ausbeute für verschiedene Reaktionstemperaturen und -zeiten ermittelt. Entsprechend wurden für die Olefine 67 und 66 der Diastereomerenüberschuß und die Ausbeute bestimmt. Außerdem wurde der Einfluß der Konzentration der Phosphonate 52 und 21, der Zusätze (LiCl, TMEDA, ZnCl2) und des Lösungsmittels (THF, Toluol) auf das Reaktionsergebnis untersucht. Das für das jeweilige Phosphonat beste Resultat ist in Abb. 73 angegeben.
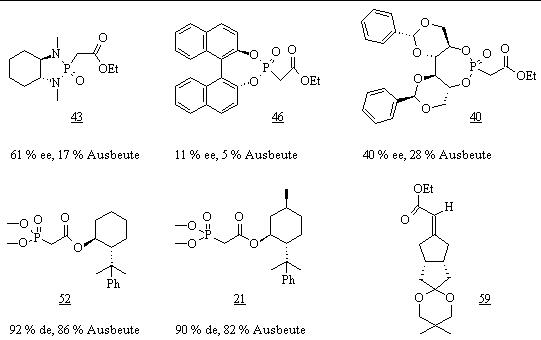
Abb. 73: Chirale Phosphonate zur HWE-Reaktion am Monoketon 29 und dabei erhaltener Enantiomerenüberschuß bzw. Diastereomerenüberschuß.
Zur Untersuchung der asymmetrischen Deprotonierung stellte man 5 chirale Amine her, mit denen man zuerst am Monoketon 29 – als Modellverbindung für Keton 72 – unter verschiedenen Reaktionsbedingungen die Reaktion optimierte (Abb. 74). Nach der Deprotonierung erhielt man durch Abfangreaktion mit Formaldehyd (monomer) das b-Hydroxyketon 108. Durch Variation der Reihenfolge, der Konzentrationen und der Zeitabstände bei der Zugabe der Reagenzien zum Monoketon 29 (Base, Zusätze, Formaldehyd), der Veränderung der Zusätze (LiCl, LiBr) und der Reaktionstemperatur konnte man den Einfluß der einzelnen Reaktionsparameter bestimmen. Außerdem ließ sich die Enantioselektivität der asymmetrischen Deprotonierung eindeutig auf eine kinetische Steuerung zurückführen. Die bei der Darstellung des Hydroxyketons 108 jeweils besten Ergebnisse der Basen 79, 85, 84, ent-36 und 19, die für den Einsatz in die Deprotonierungsreaktion zuvor mit BuLi lithiiert wurden, sind in Abb. 74 aufgeführt.
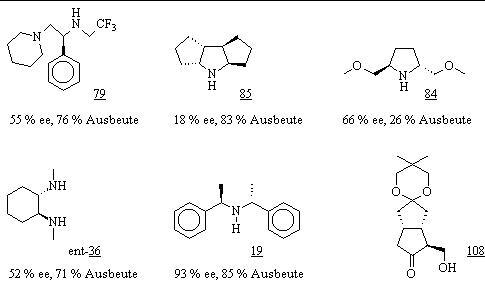
Abb. 74: Chirale Amine zur asymmetrischen Deprotonierung des Monoketons 29 und der jeweilige, erhaltene Enantiomerenüberschuß des b-Hydroxyketons 108.
Die optimierten Reaktionsbedingungen wurden auf die Totalsynthese übertragen. Dabei lief die HWE-Reaktion mit dem 8-Phenylnormentholphosphonat 52 mit einem de-Wert von 91 %, aber die asymmetrische Deprotonierung mit dem Bis(phenylethyl)amin 19 nicht wie erwartet mit einem de-Wert von 90 % sondern nur 82 % ab, was auf eine erhöhte Wärmetönung bei der Lithiumamidzugabe aufgrund des vergrößerten Reaktionsansatzes zurückzuführen ist.
Durch Abtrennen der Diastereomeren ließ sich das 3-Oxacarbacyclin (28) aber diastereomerenrein mit einem ee-Wert von > 99.4 % darstellen. Die Absolutkonfiguration von 28 stimmt mit der vom Prostacyclin (1) überein und wurde durch Drehwertvergleich einer Zwischenstufe der Totalsynthese bestätigt.
Parallel zum Syntheseverlauf, beginnend mit der a-Seitenkette, wurde die Synthese von 3-Oxacarbacyclin (28), beginnend mit der w-Seitenkette, untersucht (Abb. 75). Aufgrund der Stabilität der Ketalschutzgruppe wurde die selektive Oxidation des Diols 113 mit TEMPO (115) im Gegensatz zum Diol 112 mit befriedigender Ausbeute durchgeführt. Die lediglich 5 Syntheseschritte – ausgehend vom Monoketon 29 – zur Einführung der w-Seitenkette ließen sich präparativ in 3 Eintopfreaktionen zusammenfassen. Der anschließende Aufbau der a-Seitenkette wurde von Vaulont untersucht.
Auf diesem alternativen Syntheseweg werden Schutzgruppenoperationen auf ein Minimum begrenzt, wodurch der Syntheseaufwand verringert und die Ausbeute gesteigert werden kann. Die erhöhten Verluste durch schlechtere Ausbeuten bei asymmetrischer Deprotonierung mit anschließender Aldolreaktion und selektiver Oxidation sind am Anfang der Synthese leichter als nach Einführung der a-Seitenkette zu akzeptieren. Insgesamt sollte diese Syntheseroute deshalb ein interessanter Gegenvorschlag zur oben beschriebenen Totalsynthese sein, so daß weitere Optimierungen wünschenswert erscheinen.
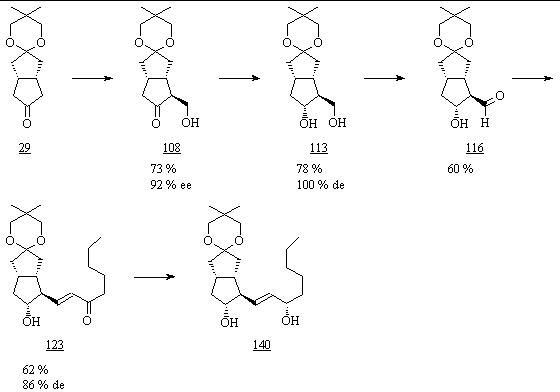
Abb. 75: Einführung der w-Seitenkette zur alternativen Totalsynthese von 3-Oxacarbacyclin (28).